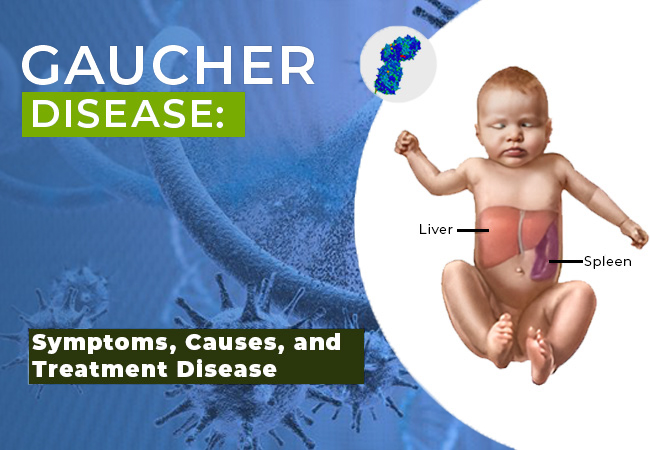
Gaucher’s disease is an uncommon genetic disorder. It is caused by a missing or deficient enzyme known as glucocerebrosidase. This enzyme helps break down fats in the body. It is responsible for breaking the fats in the body. When this enzyme is absent, fatty materials, or lipids, commence to accumulate in the liver, spleen, brain, and other organs. These can lead to various health complications.
In India, Gaucher’s disease rarely occurs with very limited cases being reported every year. However, the data available is vague and kind of uncertain.
What Is Gaucher’s Disease?
Gaucher’s disease is a hereditary condition caused by the deficiency of an enzyme. The enzyme called glucocerebrosidase, splits fats into simpler fat molecules. There is a metabolic process where the fats are transformed into energy. If this enzyme is not present, the fats stay in the cells and build up in the body. This fat accumulation does negatively affect the organs because they are not able to perform their tasks as expected. The organs that are affected the most are the brain, bone marrow, liver, lungs, and spleen.
The fats also get stored in macrophages which are a group of white blood cells. This alters the body’s waste removal system on a cellular level and leads to more destruction. Gaucher’s disease is one of the several lysosomal storage disorders (LSDs). Some more diseases in this group are Pompe, Fabry, and Hurler diseases.
Symptoms of Gaucher’s Disease:
Gaucher disease has three major categories. The major signs and symptoms of the disease are bone disease, and low hematocrit count together with low platelet count which, not everyone will have the same symptoms.
Splenomegaly or splenic hypertrophy refers to the most typical symptom which is the enlargement of the spleen. It usually goes unnoticed by the patient, but can also lead to some abdominal distension, fullness, and discomfort.
It also can lead to dysfunction of platelet and red blood cells because the spleen is a central organ for these cells. When there are low levels of circulating platelet and red blood cells, the patient experiences fatigue, easy bruising, and bleeding tendencies.
Type 1:
This is the most widespread type, constituting about 9 of 10 cases. Its symptoms are milder as compared to the others, and the brain remains unaffected. Each individual has a different level of onset and severity of symptoms. The condition can also be diagnosed in children and adults alike.
Signs and symptoms include:
- anemia
- fatigue
- lack of energy
- low blood platelet
- numbers
- nosebleeds
- osteoporosis
- bone pain
- bone thinning
- widening of bones above
- the knee joint
- damage to joints (hips and shoulders)
- liver damage
- enlarged liver
- yellow spots in the eyes
- Other complications include:
- enlarged spleen
- delayed puberty
- lung problems
- If a person has a low
- platelet count, then it is
- easier to get bruised and
- bleed because the blood
- takes a longer time to clot.
Type 2:
Type 2 is a rather severe and strange type of Gaucher’s disease. The infant type has the same symptoms as type 1 but additionally inflicts the nervous system. Also called the acute infantile neuronopathic form, it forms about twenty percent of the cases of this illness. Other signs include the presence of serious abnormalities in the brainstem and the rapid progression of severe brain damage. Symptoms start manifesting between 3 and 6 months.
In addition to the signs and symptoms seen in type 1, type 2 may involve:
- poor development
- apnea
- seizures
- jerking movements
- rigidity
- difficulty sucking and swallowing
This is a fatal form of Gaucher’s disease, and most children do not live past age 2 years.
Type 3:
This form is less common than type 1, but more prevalent than type 2. Signs and symptoms become evident in childhood and during teenage years and they are more gradual relative to type 2. It is often referred to as chronic neuronopathic Gaucher disease. Brain damage can happen, although not as severely as type 2.
In addition to the signs and symptoms of type 1, type 3 may involve:
- seizures
- dementia
- convulsions
- muscle twitches
- poor coordination
- abnormal eye movements
- delayed cognitive
- development
Norbottnian Gaucher’s Disease is a kind of type 3. Symptoms may become evident later in young adulthood.
Perinatal lethal Gaucher’s disease is regarded as the most severe type. Some complications arise before birth or during infancy. Symptoms include severe swelling, skin problems, and neurological issues. It is usually deadly before birth or a few days after.
Causes of Gaucher’s Disease:
Gaucher’s disease is a genetic condition caused by alterations in the GBA gene found on chromosome 1. The genetic condition is recessive which means an individual has to inherit a defective copy of the gene from both parents for the condition to express itself.
- An individual who possesses a single defective copy of the GBA gene from one parent is considered a carrier. A carrier is not affected by the condition but the next generation may inherit the defective copy of the gene.
- When both the parents are carriers, the child has:
- 25% chance of developing Gaucher’s disease.
- 50% chance of being a carrier.
- 25% chance of inheriting no faulty genes.
Diagnosis of Gaucher’s Disease:
Specialists use the following methods to ascertain the presence of Gaucher’s disease in a person:
- Blood Tests: These detect low levels of an enzyme called glucocerebrosidase.
- Genetic Testing: This detects variation in the GBA gene which may not be extensively identifiable.
- Prenatal Testing: Families known to carry Gaucher’s disease may have an unborn child tested for the disease through amniocentesis or chorionic villus sampling (CVS).
Blood enzyme and genetic tests done jointly provide a more definitive confirmatory diagnosis.
Treatment Options:
Treatment of Gaucher’s disease can help decrease symptoms as well as greatly enhance the quality of life. Patients suffering from type 2 Gaucher’s can take enzyme replacement therapy, but it, unfortunately, does not treat the severe brain damage rot that is almost bound to happen. Mild type 1 patients do not require treatment, though they need to be looked after more often than not. Type 1 or 3 Gaucher’s patients can benefit from more therapies.
- Enzyme Replacement Therapy (ERT): A synthetic enzyme is use to substitute the missing enzyme in the body during this therapy . The procedure is performed via a vein. ERT is effective in decreasing spleen and liver size, enhancing bone density, and raising blood cell count. However, there are no improvements in the types 2 and 3 brain symptoms.
- Substrate Reduction Therapy (SRT): SRT inhibits the formation of fatty materials deposited within the cells. Miglustat (Zavesca) is an oral therapeutic drug approved for mild to moderate type 1 Gaucher’s disease. SRT is recommended for those who cannot use ERT.
- Bone Marrow Transplant: The damaged bone marrow is substituted with bone marrow stem cells. This is seldom performed because of the possibility of complications which include infections or rejection.
Complications of Gaucher’s Disease:
Without treatment, Gaucher’s disease may result in:
- Bone breakage and joint injuries due to osteoporosis.
- Possible liver problems including cancer, but only in extreme instances.
- Research shows the possibility of a connection between neurodegenerative illness and Gaucher’s so it is reasonable to assume the patient will develop Parkinson’s.
- Calcification of cardiac ventricle valves may be more prominent in type 3.
It may be more severe with type 2, where the patient may face problems with voluntary movement, eating, and drinking, and may even get seizures.
Outlook for Gaucher’s Disease:
The life expectancy for type 1 Gaucher’s disease is close to normal with early diagnosis and treatment. However, untreated cases can lead to severe complications.
Type 1: Patients can live into their late 60s or beyond with proper care.
Type 2: This form is severe, and most patients do not survive beyond age 2.
Type 3: With treatment, patients may live into their 50s.
Final Thoughts:
Gaucher’s disease is a serious hereditary enzyme deficiency, but treatment can greatly improve the quality of life. Early diagnosis is important for managing symptoms and preventing complications. To buy Miglustat (Zavesca) in India, contact the Indian Pharma Network (IPN) for reliable and legal access in India. Early intervention, regular monitoring, and proper treatment can help patients lead longer and healthier lives.
Where can I buy Miglustat (Zavesca) in India for Gaucher’s disease treatment?
You can buy Miglustat (Zavesca) in India through authorized suppliers like Indian Pharma Network (IPN), which ensures legal access to life-saving medications. It is essential to procure the drug from licensed pharmaceutical suppliers to ensure authenticity and compliance with regulatory standards.
Who is the best Miglustat (Zavesca) importer in India?
Indian Pharma Network (IPN) is a trusted Miglustat (Zavesca) importer in India. We offer a secure and compliant pathway to access this medication. IPN works with global manufacturers and authorized distributors to source the drug legally for patients who require treatment for Gaucher’s disease.
Can I buy Miglustat (Zavesca) in SAARC countries?
Yes, patients and healthcare providers can buy Miglustat (Zavesca) in SAARC countries, including Bangladesh, Nepal, Bhutan, Sri Lanka, the Maldives, Pakistan, and Afghanistan, through licensed pharmaceutical suppliers. It is crucial to ensure regulatory approvals in each country before importing the medication.
Who is a reliable Miglustat (Zavesca) supplier in India for hospitals and distributors?
Indian Pharma Network (IPN) is a well-established Miglustat (Zavesca) supplier in India. We cater to hospitals, facilitators, and individual patients. IPN ensures that the medicine is sourced from genuine and approved manufacturers while complying with Indian pharmaceutical regulations.
Where can I buy Miglustat (Zavesca) in major Indian cities like Lucknow, Delhi, Noida, and Gurgaon?
Patients can buy Miglustat (Zavesca) in Lucknow, Delhi, Noida, Gurgaon, as well as in other key cities such as Mumbai, Chennai, Hyderabad, Kolkata, Pune, Ahmedabad, Bangalore, Chandigarh, and Jaipur. We help facilitate nationwide delivery of this medication while ensuring compliance with import and pharmaceutical regulations.